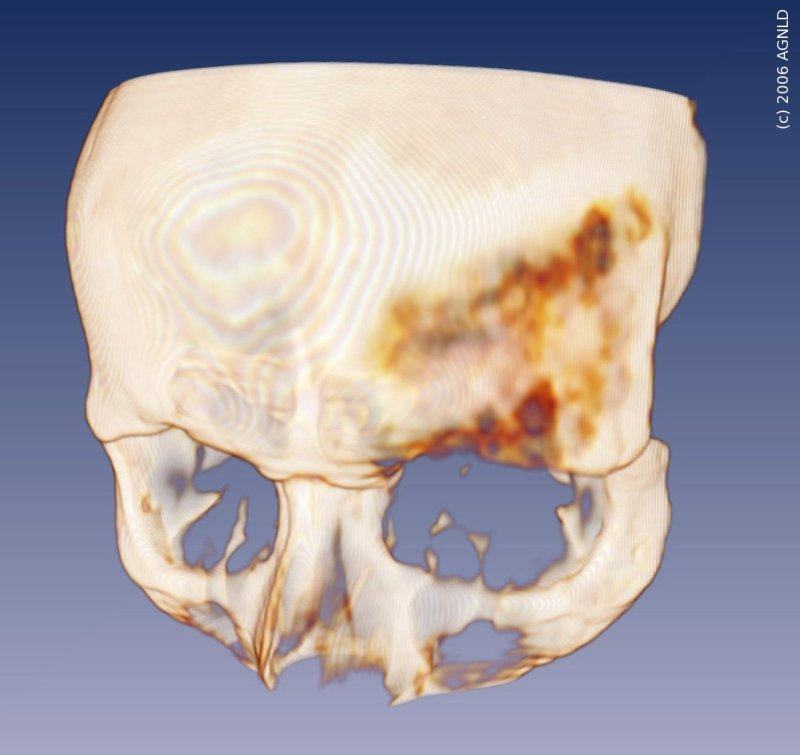
Fibröse Dysplasie am Stirnbein (Os frontale). Die rötliche Färbung zeigt deutlich die Verringerung der Mineralisierung des Knochens. [Info]
| home | cave | literatur | links | login | impressum |
Fibröse Dysplasie (FD) ist zwar eine relativ seltene Erkrankung der Knochen, jedoch die häufigste Knochenfehlbildung, die im Kindes- und Jugendalter auftritt. Dabei handelt es sich um eine chronische Störung im Aufbau von neuer Knochenmasse. Statt strukturiertem und mineralisiertem neuen Knochen wird nur Bindegewebe gebildet. In einer Röntgenaufnahme ist ein milchglasartiges Erscheinungsbild typisch für betroffene Knochen. Häufig kommt es durch FD zu einer Schwächung des Knochens, verbunden mit gestiegenem Frakturrisiko, sowie starken Schmerzen. Außerdem können Verformungen des Knochens auftreten. Im Schädelbereich können durch das ungeordnete Knochenwachstum Nervenbahnen (Seh- oder Hörnerv) oder das Gehirn beeinträchtigt werden, was dann in selteneren Fällen zu physiologischen Beeinträchtigungen oder gar Ausfällen führen kann.
FD kann einen einzelnen Knochen (monostotisch) oder mehrere Knochen (polyostotisch) betreffen. Die monostotische FD tritt häufiger auf als eine polyostotische FD. Gesichts- und Schädelknochen sind am häufigsten betroffen. Problematisch sind Erkrankungen der Extremitäten, wie Oberschenkel, Oberschenkelhals, Unterarm etc., da sie besonders stark belastet und damit anfällig für Brüche sind. Weiterhin können Erkrankungen an Rippen und auch Wirbelkörpern auftreten. Wenn einmal alle betroffenen Knochen festgestellt wurden (z. B. über eine Knochenszintigraphie), wäre es sehr ungewöhnlich, wenn später neue Knochen betroffen sind (evtl. Verwechslung mit Morbus Paget).
Oft wird eine FD nur per Zufall diagnostiziert, z. B. wenn Knochen bereits unter kleiner Belastung brechen oder bei einer CT-Untersuchung. Eine Erkrankung im Kindes- oder Jugendalter wird jedoch meist recht schnell erkannt. Neben häufigen Knochenbrüchen äußert sich eine FD auch in einer Störung des Hormonhaushalts (Überschuß an Wachstumshormonen), weshalb an FD erkrankten Heranwachsenden oft ein beschleunigter Reifungsprozeß beobachtet werden kann. Es können auch Pigmentstörungen der Haut ("Café-au-lait-Flecken") oder auch starke Verformungen der Kieferknochen (Cherubismus), verbunden mit den bereits erwähnten hormonellen Störungen (Cushing-Syndrom, McCune-Aulbright-Syndrom) auftreten. Eine "einfache" (und zumeist monostotische, aber auch polyostotische) FD wird manchmal auch nach dessen Erforschern als Jaffé-Lichtenstein-Syndrom bezeichnet. Jedoch handelt es sich bei FD, McCune-Aulbright-Syndrom und auch Jaffé-Lichtenstein-Syndrom um die gleiche ursächliche Erkrankung. Seltener kann eine FD gemeinsam mit Myxomen der Skelettmuskulatur (Mazabraud-Syndrom) oder Fehlfunktionen von Herz, Leber, Bauchspeicheldrüse, Schilddrüse oder anderen Organen auftreten. Durch den verstärkten Knochenumbauprozeß ist eine FD oft mit erhöhten Werten der alkalischen Phosphatase (AP) im Blut und Desoxypyridinolin (DPD) im Urin verbunden (was aber umgedreht nicht gleich eine Erkrankung an FD bedeuten muß). Daher sollten diese Werte bei FD-Betroffenen regelmäßig untersucht werden, um Veränderungen in der Aktivität der FD festzustellen.
Im Zusammenhang mit FD können sich Aneurysmatische Knochenzysten oder Solitäre Knochenzysten in dem betroffenen Knochen entwickeln. Derartige Knochenzysten sind gutartige, jedoch schnellwachsende und das Knochenmaterial schädigende, meist blutgefüllte, schmerzhafte Läsionen (Schädigungen), die in der Regel operativ entfernt werden müssen.
FD wird durch eine nichtvererbbare Mutation des sogenannten G-Proteins ausgelöst (im α-subunit). Die Mutation befindet sich im Gen GNAS (oder auch Gsα-Gen) des 20. Chromosoms. G-Proteine sind im Zell-Stoffwechsel für die Signalweiterleitung extrem wichtig. Durch die Mutation kommt es zu einer Überproduktion des Enzyms Adenylylcyclase, das die Katalyse von ATP zu cAMP steuert (das ist dann der eigentliche Signalübertragunsprozess). cAMP regelt z. B. die Herzfrequenz, Relaxation der glatten Muskulatur, die Wirkung von zahlreichen Hormonen und eben auch die Knochenzellen, die für den Aufbau von Knochen verantwortlich sind (Osteoblasten). Der Grund für diese Mutation ist noch unbekannt. Sie kann bereits im Föten-Stadium während der Schwangerschaft auftreten (dann treten die Symptome bereits im Kindes- und Jugendalter auf) oder auch erst später, nach der Geburt. Da FD sehr stark mit Hormon- und anderen Zellstoffwechselprozessen verbunden ist, kann eine FD nach der Pubertät aufhören. Hormonpräparate (wie z. B. die Pille) oder eine Schwangerschaft können einen positiven oder auch negativen Einfluß auf die Weiterentwicklung der FD haben.
Der Gendefekt betrifft nur Körperzellen, nicht die Keimzellen (das erste nennt man dann somatische Mutation, dagegen bei Keimzellen gametische Mutation). D. h., der Gendefekt kann nicht an die Nachkommen weitergegeben werden. Findet die Mutation, die FD auslöst, während der frühen embryonalen Entwicklung in der Zellmasse statt, kommt es vermutlich zum McCune-Albright-Syndrom; Mutation zu einem späteren Zeitpunkt der embryonalen Entwicklung löst vermutlich die polyostotische FD aus. Und eine Mutation nach der Geburt (im Kindes- oder gar Erwachsenenalter) wird für die monostoische FD verantwortlich gemacht. Je nachdem, wo die Mutation in der Zellmasse während der embryonalen Entwicklung stattfindet, gibt es später in diesen Körperregionen die Krankheit.
Die Keimzellen entstehen aus Urkeimzellen, die bereits während des Embryowachstums von den Körperzellen getrennt werden (und dann in der Keimdrüse gelagert werden). Eine Vererbung von FD ist daher nahezu ausgeschlossen.
Eine FD ist nicht heilbar. Jedoch können neben operativen Eingriffen zur Entfernung von betroffenem Knochenmaterial oder zur Stabilisierung auch Chemotherapien mit Bisphosphonaten angewendet werden. Es gibt verschiedene Klassen von Bisphosphonaten, die alle die Wirkung der knochenabbauenden Zellen (Osteoclasten) einschränken, und damit die FD verzögern und auch Schmerzen lindern können. Am häufigsten finden Pamidronate (z. B. Aredia), Risedronate (z. B. Actonel) und Zoledronate (z. B. Zometa) Anwendung.
Obwohl es teilweise erhebliche physische Einschränkungen bei FD-Patienten gibt (z. B. häufige Operationen oder Deformationen), so wurde jedoch im Rahmen einer Studie festgestellt, daß in allen wichtigen Bereichen des Lebens, wie soziale Beziehungen, Bildung und Karriere, die Lebensqualität völlig normal ist. Dagegen leiden Eltern von Kindern, die an FD erkrankt sind, of sehr stark unter der Erkrankung. Es sei daher wichtig, die Eltern über die Krankheit aufzuklären und zu versichern, daß Lebenserwartung sowie soziale und emotionale Gesundheit der Kinder nicht beeinträchtigt ist.
schauen Sie auch unter
Wikipedia
ALL RIGHTS RESERVED
© 2005-2008 NONLINEAR DYNAMICS GROUP (AGNLD), UNIVERSITY OF POTSDAM.
© 2008-2024 TRANSDISZIPLINÄRE KONZEPTE UND METHODEN, POTSDAM-INSTITUT FÜR KLIMAFOLGENFORSCHUNG (PIK).